|
|
MitoTIP
An in silico tool for predicting pathogenicity of novel mitochondrial tRNA variants, developed by Neal Sondheimer and Sanjay Sonney (http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005867). The pathogenicity scoring from MitoTIP is intended as a starting point for analysis of newly observed tRNA variants. It is based only on database frequencies, the nature of the nucleotide change (transversion/transition/deletion), and conservation scoring. No clinical histories, heteroplasmy data, or functional studies are included. Variants must be further evaluated by the end user in the context of the actual patient heteroplasmy, and the heteroplasmy seen in affected and unaffected family members. Other additional lines of evidence are suggested below. MitoTIP integrates multiple sources of information to provide a prediction for the likelihood that novel single nucleotide variants or deletions in tRNA-encoding sequences would cause disease. The sources of information used include:- GenBank deposited mitochondrial sequence
- Annotations of pathogenicity from MITOMAP
- Conservation across species
- The position of the variant within the tRNA
- Whether the nucleotide change is a transition or transversion
| Current Raw Scores | Quartile | Quartile Ranking / |
| >16.25 | >75 - 100% | likely pathogenic |
| 12.66 – 16.25 | >50 - 75% | possibly pathogenic |
| 8.44 – 12.66 | >25 - 50% | possibly benign |
| < 8.44 | 0 - 25% | likely benign |
New Additional Features
MitoTIP sub-scoring details may be found on the details page for each tRNA variant. The MitoTIP percentile score provides the link to the details page. For example, in the screenshot below, clicking on the percentile score of 22.5% for variant 5555G, will take you to its details page. Variant frequencies and top haplogroups are now shown on a variant's details page. All haplogroups with a variant frequency >10% are listed. If no haplogroup has a variant frequency >10%, then the top haplogroup is listed.A "Frequency Alert" asterisk is added to the MitoTIP ranking if a variant is found (1) at >1% in at least one of the macro-lineages L, M, or N, or (2) at >10% in at least one of the major (single letter) haplogroups, with minimum # sequences = 10. Note that the frequency alert is for information purposes only. It does not confirm a benign status, especially if a variant is found outside of the high-frequency haplogroup.
An indication of Mitomap’s “confirmed pathogenic” status replaces the interpretation for those specific tRNA mutations in the variant summary, but the raw MitoTIP score may be found on variant details page.
Lines of Evidence Recommended
Additional lines of evidence should be pursued to confirm pathogenicity for any tRNA variant, regardless of its MitoTip score: determination of heteroplasmy levels, correlation of heteroplasmy with phenotype, functional cybrid studies, tRNA steady state levels, single fiber studies.
Screenshot of Mitomaster query details showing the MitoTIP prediction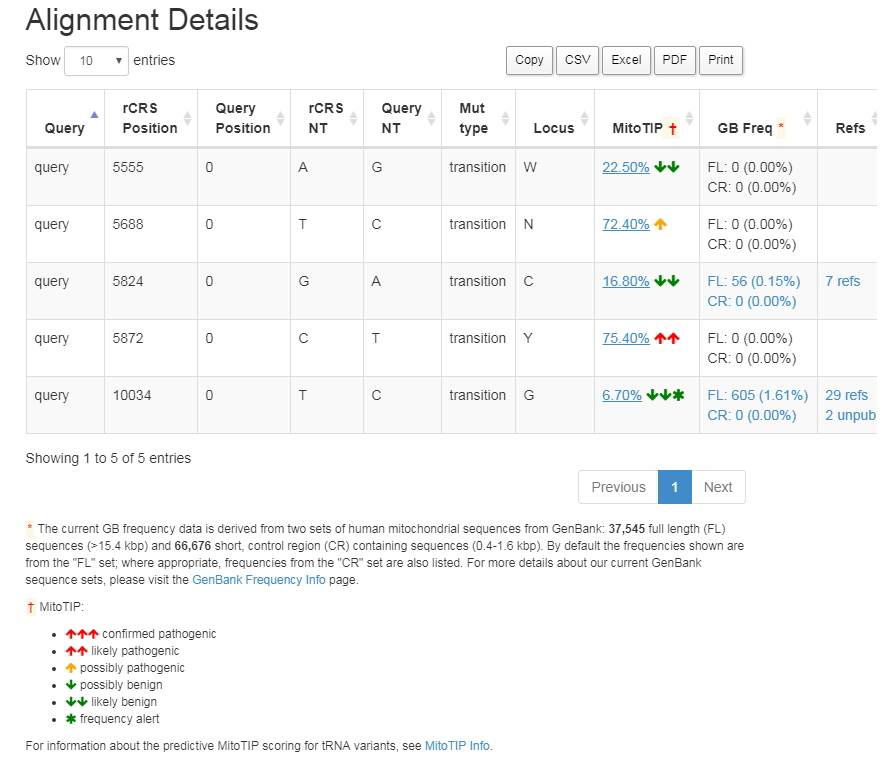 |
Users can input any tRNA-encoding position into MITOMAP’s allele search or into MITOMASTER’s SNV Query and retrieve the predicted pathogenicity score of any possible change at that position. Any variant whose status is not already confirmed as pathogenic will be displayed with a prediction (likely benign/possibly benign/possibly pathogenic/likely pathogenic) derived from quartiles of the pathogenicity score. The use of this platform will simultaneously direct users to underlying literature supporting the assignment of confirmed or suspected mutations.
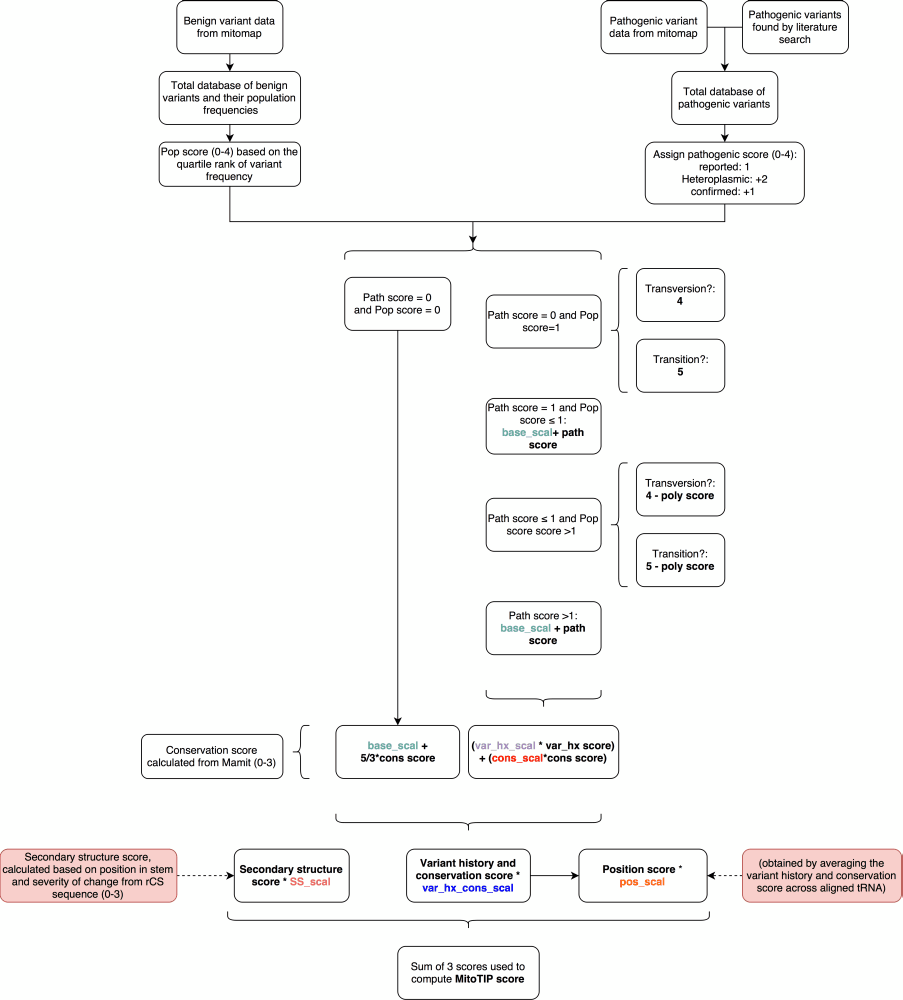
Weaknesses of this system include its reliance on databases, which may fall out of date, contain errors or have sequence entries from patients with mitochondrial disorders. The underlying calls of pathogenicity represent a best effort at identifying all legitimate reports of mitochondrial variants but may have missed some reports and may include other reports that inaccurately ascribe pathogenicity to neutral variants. Providing MitoTIP data in the context of other information will allow users to integrate multiple sources of information when assessing unfamiliar variants. The system’s strength is also in its use of databases as we can easily update it with new information incorporated into MITOMAP so that the system will improve in sensitivity and specificity over time. The system is designed for the evaluation of novel variants, where previous data confirming pathogenicity is unavailable. Indeed, known pathogenic mutations such as mt.8344A>G score poorly in MitoTIP because the position is neither well conserved nor in a secondary structure location commonly associated with disease. We also have not incorporated the user-detected heteroplasmy of a query variant into our scoring. It is widely accepted that heteroplasmic variants are more likely to be pathogenic and low penetrance variants that are homoplasmic are less common. The pathogenicity scoring from MitoTIP for newly observed variants can and should be evaluated by the end user in the context of the actual patient heteroplasmy, and the heteroplasmy seen in affected and unaffected family members.Scoring Flowchart
Ideas, requests, problems regarding Foswiki? Send feedback